2Uniform Manifold Approximation and Projection for Dimension Reduction (UMAP)
If you have not read the t-SNE, I strongly suggest to scan that section before you continue with UMAP.
Similar to t-SNE, UMAP is used to reduced the dimensions of data. Despite that it is slighlt better in preserving the global structure, I would put in the same class as t-SNE that is the main focus is to give priority to accurately reflecting the relationships of nearby points. So the idea is the same. We want to reduce the dimension of the data to a set of new dimesion so that we can see the relationship between the data points. We are interested in tightly clustered data points because we think they are showing a stronger or more meaningful relationship in the original high-dimensional space. These tight clusters likely represent similar or related data points, and by preserving the local structure, we aim to retain these key relationships in the lower-dimensional representation. This helps in identifying patterns, trends, or groupings that are otherwise difficult to visualize. While global relationships are not entirely ignored, the emphasis on local neighborhoods ensures that small but significant clusters are faithfully represented, which is particularly useful in tasks like clustering, classification, or understanding complex datasets.
Later we are going to see exactly how UMAP works but for now let’s consider it as a tool that takes the input data, transforms it and gives us some output. We are going to see how to use UMAP in R.
3 UMAP in R
There is a few good implementation on UMAP in R. We are going to use uwot package. It should work on most platforms. An alternative would be umap package uwot gives more flexebility in terms of functions it can perform and parameters etc.
we are going to use the same data that we have been using for t-SNE
Code
# Visualize the original data in 3D, distinguishing clusters and subclusterslibrary(dplyr)library(plotly) # For interactive 3D plots# Set seed for reproducibilityset.seed(123)# Number of points per subclustern <-25# Manually define main cluster centers (global structure)main_cluster_centers <-data.frame(x =c(0, 5, 10, 15),y =c(0, 5, 10, 15),z =c(0, 5, 10, 15),cluster =factor(1:4))# Manually define subcluster offsets relative to each main cluster# These small offsets will determine subcluster locations within each main clustersubcluster_offsets <-data.frame(x_offset =c(-0.25, 0.25, -0.25, 0.25),y_offset =c(-0.25, -0.25, 0.25, 0.25),z_offset =c(0.25, -0.25, -0.25, 0.25),subcluster =factor(1:4))# Initialize an empty data frame to hold all datadata <-data.frame()# Generate data for each main cluster with subclustersfor (i in1:nrow(main_cluster_centers)) {for (j in1:nrow(subcluster_offsets)) {# Calculate subcluster center by adding the offset to the main cluster center subcluster_center <- main_cluster_centers[i, 1:3] + subcluster_offsets[j, 1:3]# Generate points for each subcluster with a small spread (to form local clusters) subcluster_data <-data.frame(gene1 =rnorm(n, mean = subcluster_center$x, sd =0.25), # Small spread within subclustersgene2 =rnorm(n, mean = subcluster_center$y, sd =0.25),gene3 =rnorm(n, mean = subcluster_center$z, sd =0.25),cluster = main_cluster_centers$cluster[i],subcluster = subcluster_offsets$subcluster[j] )# Add generated subcluster data to the main data frame data <-rbind(data, subcluster_data) }}plot_ly( data, x =~gene1, y =~gene2, z =~gene3, color =~cluster,symbol=~subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter3d", mode ="markers",size =5) %>%layout(title ="Original 3D Data with Clusters and Subclusters",scene =list(camera =list(eye =list(x =0.3, y =2.5, z =1.2) # Change x, y, z to adjust the starting angle ) ) )
This data has just three dimentions for 400 samples (4 major clusters within each we have subclusters). We are now going to do a UMAP (using umap function) on this data and see the results. We also do t-SNE and show the results side by side. There are parameters to set but we just go for the default ones except for n_neighbors in UMAP which we set to 30 to be similar to t-SNE.
Code
library(Rtsne)library(uwot)set.seed(123)tsne_results <-Rtsne(as.matrix(data[, c("gene1", "gene2", "gene3")]),perplexity =30,)set.seed(123)umap_results <-umap(as.matrix(data[, c("gene1", "gene2", "gene3")]),n_neighbors =30,)tsne_plot <-plot_ly(as.data.frame(tsne_results$Y), x =~V1, y =~V2, color =~data$cluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="" )umap_plot <-plot_ly(as.data.frame(umap_results), x =~V1, y =~V2, color =~data$cluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="" )subplot(tsne_plot%>%layout(showlegend =FALSE), umap_plot%>%layout(showlegend =FALSE),titleX = T,titleY = T,margin =0.2)%>%layout(annotations =list(list(x =0.13 , y =1.035, text ="t-SNE", showarrow = F, xref='paper', yref='paper'),list(x =0.85 , y =1.035, text ="UMAP", showarrow = F, xref='paper', yref='paper')))
What we see here is lower dimension repsentation of our data (from 3D to 2D). Both methods did a great job (as expected!) to capture the clusters. We are going to leave t-SNE for now and only focus on how we can use UMAP and what we can do with it.
3.1 Most important parameters
This implementation of UMAP actually offers many parameters to configure. Fortunately, most of them can be left at their default values. However, to get started, there are three key parameters that we need to focus on for now:
X: This represents the input data that we want to reduce in dimensionality. It can be any numerical dataset (or factor, more on this later), such as a matrix or data frame in R.
n_components: The dimension of the space to transform the original data into.This is basically, the size of the lower dimension.
n_neighbors: This parameter controls the size of the local neighborhood that UMAP uses for each point. It essentially determines how many nearby points influence the embedding of a given point, with smaller values focusing more on local structure and larger values considering broader relationships.
We can run the function by calling the umap(data,n_components=2,n_neighbors=10). Remember to set the seed for reproducibility.
The n_neighbors parameter is the most important here. We can visualize how it affects the outcome of dimensionality reduction.
Code
umap_data_frames<-c()for(nn inc(5,10,15,20,30,50,100,200,300,400)){set.seed(123)umap_results <-umap(as.matrix(data[, c("gene1", "gene2", "gene3")]),n_neighbors = nn,) umap_data_frames<-rbind(umap_data_frames,data.frame(umap_results,data[,c(4,5)],n_neighbors=nn))}umap_plot<-plot_ly( umap_data_frames, x =~X1, y =~X2, color =~subcluster,frame=~n_neighbors,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="" )umap_plot
Here i have plotted the data and colored the sub clusters. Surprisingly, the algorithm is quite robust when it comes to little changes to n_neighbors parameters but as we use larger number of neighbors the algorithm starts capturing more of a global structure than local. This is similar behavior to t-SNE but is less sensitive to the changes.
There are a few other parameters to play with, namely spread and min_dist. We are going to talk a bit about them later in the math section but for now let’s give you an intiutaive explanation for them. The spread parameter sets how much “space” the UMAP algorithm has to distribute points across the lower-dimensional space. A higher value of spread allows points to be distributed more widely, meaning that the embedding will appear more “spread out.” Conversely, a lower spread value will make the points more tightly packed together across the entire embedding. min_dist on the other hand, sets how close points can be to each other. Smaller min_dist values lead to tighter local clusters in the embedding. Obviousyl, this has to be set relative to spread.
For an example let’s focus on one our highest n_neighbors results (n_neighbors=200).
Code
umap_data_frames<-c()for(spr inc(1:10)){set.seed(123)umap_results <-umap(as.matrix(data[, c("gene1", "gene2", "gene3")]),n_neighbors =200,min_dist = spr/100,spread = spr) umap_data_frames<-rbind(umap_data_frames,data.frame(umap_results,data[,c(4,5)],spread=spr))}umap_plot<-plot_ly( umap_data_frames, x =~X1, y =~X2, color =~subcluster,frame=~spread,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="" )umap_plot
As you can can see UMAP changes the scales of how the points are spreads out the points in the lower dimension. We can now see the effect of min_dist:
Code
umap_data_frames<-c()for(mn_dst in1:8){set.seed(123)umap_results <-umap(as.matrix(data[, c("gene1", "gene2", "gene3")]),n_neighbors =200,min_dist = mn_dst,spread =10) umap_data_frames<-rbind(umap_data_frames,data.frame(umap_results,data[,c(4,5)],min_dist=mn_dst))}umap_plot<-plot_ly( umap_data_frames, x =~X1, y =~X2, color =~subcluster,frame=~min_dist,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="" )umap_plot
In this plot instead of umap “streaching out” the space, the distance between the points are now increasing. Although it might be a bit confusing, you can think of as if spread focuses on global structure whereas min_dist focuses on local relationships. In any case there is no best value for these parameters. One has to experiment with them to get an desired output.
3.2 Mixed data types
Sometimes it is possible to encounter mixed data types, combinations of numeric, categorical, and even binary data. This implementation of UMAP offers flexibility for handling such datasets through its metric parameter. It can handle multiple types of distance metrics simultaneously, meaning we can tune the way UMAP processes different subsets of our data based on their characteristics. For instance, we might use Euclidean distance for continuous variables, Hamming distance for binary data, or categorical distance for factor variables.
These are basically the distances that are supported now:
Euclidean: Best suited for continuous numeric data.
Cosine: Useful when you want to measure the angle between vectors (common in text or word embedding tasks).
Manhattan: Focuses on absolute differences between values, which can be useful for certain types of numeric data.
Hamming: Ideal for binary or bit-wise data.
Categorical: Designed specifically for factor variables, ensuring that categorical data is treated appropriately.
If our dataset is a data frame or matrix with multiple data types, UMAP allows us to specify a list of metrics, each applied to specific columns. For example, we could assign Euclidean distance to a block of numeric columns and Manhattan distance to another block. In our case, we have used default parameters which is euclidean distance. Have a look at help page of umap function to see how you can incorporate different distance metric.
3.2.1 Machine learning
Despite the fact that the most common application of UMAP, at least in life sciences, is to visualize data and confirm clusters, its utility goes beyond visualization and can sometimes significantly improve machine learning workflows. The idea here is to project high-dimensional data into a lower-dimensional space and train the model on these limited space. Basically, what we want to do is to mitigate the curse of dimensionality, reduces computational costs, and eliminates noise, so we get cleaner and more compact data representations.
Training a model is streight forward, we could just take the umap latent scores and use them in the model as features. In our case, let’s see if we can use Random Forest to do that. We are going to predict the subclusters in our data using both the original and transformed features:
Using the orignal features
Code
set.seed(123)umap_results <-umap(as.matrix(data[, c("gene1", "gene2", "gene3")]),n_neighbors =10,)X <- data[, c("gene1", "gene2", "gene3")] # Predictor variables (genes)y <- data$subcluster # Target variable# Set seed for reproducibilityset.seed(123)rf_model <- randomForest::randomForest(y ~ ., data =data.frame(X, y), family = binomial)print(rf_model)
Call:
randomForest(formula = y ~ ., data = data.frame(X, y), family = binomial)
Type of random forest: classification
Number of trees: 500
No. of variables tried at each split: 1
OOB estimate of error rate: 26.25%
Confusion matrix:
1 2 3 4 class.error
1 72 14 6 8 0.28
2 8 73 9 10 0.27
3 7 11 74 8 0.26
4 6 10 8 76 0.24
The performance is OK. We can now perform the same modeling but using the UMAP scores instead of the original data.
Code
set.seed(123)umap_results <-umap(as.matrix(data[, c("gene1", "gene2", "gene3")]),n_neighbors =10,ret_model =TRUE)X <- umap_results$embedding # Get the lower dimensiony <- data$subcluster # Target variable# Set seed for reproducibilityset.seed(123)rf_model <- randomForest::randomForest(y ~ ., data =data.frame(X, y), family = binomial)print(rf_model)
Call:
randomForest(formula = y ~ ., data = data.frame(X, y), family = binomial)
Type of random forest: classification
Number of trees: 500
No. of variables tried at each split: 1
OOB estimate of error rate: 25.25%
Confusion matrix:
1 2 3 4 class.error
1 72 12 6 10 0.28
2 10 74 9 7 0.26
3 6 9 75 10 0.25
4 4 9 9 78 0.22
The performance became slightly better and we are using one less dimension. Please note that in calling the umap function, i have used ret_model = TRUE. This is not because we want to do modeling on the scores (it does not change anything) but rather because retaining the model allows us to reuse the trained UMAP model on new, unseen data. This is an important feature of UMAP, which allows us to project new data that has not been part of the original dataset onto the same lower-dimensional space. We want to do that because when testing our machine learning model, we need to have consistency between the training and testing data representations. By projecting the test data onto the same UMAP-reduced space as the training data, we ensure that the relationships and structures are preserved, making the model’s performance evaluation more reliable. This is essential for maintaining the integrity of the testing phase, as the model can only make valid predictions if the test data is represented in the same lower-dimensional space as the data it was trained on. Without this, differences in dimensionality reduction could lead to misleading results and reduced model performance.
Let’s generate some test data and see how UMAP does the projection.
Code
set.seed(123)gene1<-rnorm(20,mean =mean(data$gene1),sd =sd(data$gene1))gene2<-rnorm(20,mean =mean(data$gene2),sd =sd(data$gene2))gene3<-rnorm(20,mean =mean(data$gene3),sd =sd(data$gene3))data_test <-data.frame(gene1=gene1,gene2=gene2,gene3=gene3)plot_ly( data_test, x =~gene1, y =~gene2, z =~gene3,type ="scatter3d", mode ="markers",size =5) %>%layout(title ="Original 3D Data with Clusters and Subclusters",scene =list(camera =list(eye =list(x =0.3, y =2.5, z =1.2) # Change x, y, z to adjust the starting angle ) ) )
We are not going to measure the performance etc because we just randomly generated these points but in real application we need to do that.
So to summarize, we can use the projection capability of UMAP in order to map new unseen data points onto the latent structure and use for variety of tasks including predictive modelling etc.
3.3 Data integration
UMAP’s flexibility and ability to handle multiple data types make it an excellent tool for data integration, meaning when combining datasets from different sources or modalities. In many machine learning and bioinformatics applications, datasets may come from varied experiments or measurement techniques (e.g., RNA-seq, metabolomics, clinical data), and we might want to integrate into a common analysis space.
You need to have the same samples across the different modalities to be able to perform intergration. It is possible to modify UMAP to do integration without having corresponding samples across the data sources but this is not the standard capability of UMAP.
In order to demonstrate data integration, we need to simulate some more data. In this case, i have simulated a dataset that has six genes. There are some correlation between these genes but the key differences is that three of these are showing some form of clustering while the other three show different clusters.
Code
# Set seed for reproducibilityset.seed(123)# Number of points per subclustern <-25# Function to introduce correlation between gene1:gene3 and gene4:gene6generate_correlated_data <-function(n, main_cluster_id, subcluster_offset) {# Generate subcluster-visible data in gene1:gene3 gene1 <-rnorm(n, mean = subcluster_offset[1], sd =0.25) # Subcluster-visible in gene1:gene3 gene2 <-rnorm(n, mean = subcluster_offset[2], sd =0.25) gene3 <-rnorm(n, mean = subcluster_offset[3], sd =0.25)# Introduce correlation between gene1:gene3 and gene4:gene6 to ensure full visibility in all 6 genes gene4 <-0.8* gene1 +rnorm(n, mean = main_cluster_id *2, sd =0.25) # Main clusters visible in gene4:gene6 gene5 <-0.8* gene2 +rnorm(n, mean = main_cluster_id *2, sd =0.25) gene6 <-0.8* gene3 +rnorm(n, mean = main_cluster_id *2, sd =0.25)return(data.frame(gene1, gene2, gene3, gene4, gene5, gene6))}# Define subcluster offsets (subclusters in gene1:gene3)subcluster_offsets <-list(c(-0.25, -0.25, 0.25),c(0.25, -0.25, 0.25),c(-0.25, 0.25, -0.25),c(0.25, 0.25, -0.25))# Initialize an empty data frame to hold all datadata <-data.frame()# Generate data for each main cluster and subclusterfor (main_cluster_id in1:4) { # 4 main clustersfor (subcluster_id in1:4) { # 4 subclusters per main cluster# Generate data for the current subcluster subcluster_data <-generate_correlated_data(n, main_cluster_id, subcluster_offsets[[subcluster_id]])# Add the cluster and subcluster labels subcluster_data$main_cluster <-factor(main_cluster_id) subcluster_data$subcluster <-factor(subcluster_id)# Combine with the main data data <-rbind(data, subcluster_data) }}set.seed(123)full_model<-umap(data[,1:6], n_neighbors =30)set.seed(123)D1<-umap(data[,1:3], n_neighbors =30)set.seed(123)D2<-umap(data[,4:6], n_neighbors =30)full_data<-plot_ly(as.data.frame(full_model), x =~V1, y =~V2, color =~data$main_cluster,symbol =~data$subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5)D1_data<-plot_ly(as.data.frame(D1), x =~V1, y =~V2, color =~data$main_cluster,symbol =~data$subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5)D2_data<-plot_ly(as.data.frame(D2), x =~V1, y =~V2, color =~data$main_cluster,symbol =~data$subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5)subplot(full_data%>%layout(showlegend =FALSE), D1_data%>%layout(showlegend =FALSE), D2_data%>%layout(showlegend =FALSE),nrows =2,titleX = T,titleY = T,margin =0.03)%>%layout(annotations =list(list(x =0.13 , y =1.035, text ="Full data", showarrow = F, xref='paper', yref='paper'),list(x =0.85 , y =1.035, text ="Dataset 1", showarrow = F, xref='paper', yref='paper'),list(x =0.13 , y =0.3, text ="Dataset 2", showarrow = F, xref='paper', yref='paper')))
In the three subplots above, you see the results of UMAP on the full data (all six variables), Dataset 1 (first three variables) and Dataset 2 (last three variables). In all the plots, the colors show one type of clusters and shapes show a different type. Considering the full data, it is clear that colors are separated and within each color we have relatively good separation of shapes. However, if we do UMAP on each dataset in isolation, none of the datasets can show us a good separation of both cluster types.
Now the point here is that for some reasons we don’t want to merge our data sources. It could be because they come from different distributions, each dataset might have some hidden pattern, or we want to preserve the original structure of each dataset. In fact, in most cases, we really don’t want to do the merging and apply UMAP on the merged data directly. What we are actually interested in doing is to map each dataset to an intermediate space that preserves as much information as possible from each dataset individually, while having the exact same statistical properties.
In this case, this intermediate stage is a graph that represents similarities between the samples. similarity_graph function from uwot allows us to do such a mapping. It is important to note that similar to t-SNE, this similarity is affected by so many parameters but most importantly n_neighbors. In fact, you can think about the approach as follow, we are going to do UMAP on each of datasets in isolation, extract the similarity graph from the UMAP object and merge these similarity graphs into a single one and then only visiualize the merged one.
So let’s start creating the similarity graphs first:
We can now easily merge these graphs using simplicial_set_intersect or simplicial_set_union function. In many applications we want to use simplicial_set_intersect as it focuses on the shared structures between the datasets but if the total structure is of interest, we could use union. After building the merged similartiy we can use optimize_graph_layout function to map these similarty onto a lower dimensional space and visualize it.
set.seed(123)# Combine the two representations into oneinteg_umap <-simplicial_set_intersect(x = D1_sim, y = D2_sim)set.seed(123)umap_scores <-optimize_graph_layout(integ_umap)plot_ly(as.data.frame(umap_scores), x =~V1, y =~V2, color =~data$main_cluster,symbol =~data$subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="",showlegend =FALSE )
In the figure you see the integrated UMAP space. This acutally looks really good. But the question is can we make this a bit better by for example trying to make color clusters a bit tighter?
Fortunately, UMAP gives us weight parameter in simplicial_set_intersect function which can be used to tune relative influence each of the dataset on the final embedding. When we called the function above we assigned dataset 1 to x and dataset 2 to y. A weight of 0.5 gives equal influence. Values smaller than 0.5 put more weight on x, meaning dataset 1. Values greater than 0.5 put more weight on y, that is dataset 2.
In this specific case, sine i want tighter colors, i want to give slightly more weight to the dataset 2.
set.seed(123)# Combine the two representations into oneinteg_umap <-simplicial_set_intersect(x = D1_sim, y = D2_sim,weight =0.6)set.seed(123)umap_scores <-optimize_graph_layout(integ_umap)plot_ly(as.data.frame(umap_scores), x =~V1, y =~V2, color =~data$main_cluster,symbol =~data$subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="",showlegend =FALSE )
We can see the results, we clusters are now much tighter than before and still we have OK clustering of the shapes. Can you find a better value for the weight? This takes a bit of experimenting with weight. But you can also use different paramters when contructing the initial similarity graph. For example you might suspect that the clusters are more refined in dataset 1 vs dataset 2, then you can use smaller number of neighbors. Other parameters can also be changed depending on the specific dataset. This wraps up the data integration approach of UMAP.
3.4 Supervised
Although UMAP is an unsupervised method, it can be adapted in a way that mimics supervised learning. This doesn’t mean UMAP maps data from an input space to a specific output space. Instead, the constructed lower-dimensional space is adjusted to reflect class separability, tuning the embedding so that data points from the same class are closer together, while points from different classes are more distinct. In this case, we need some target data. This is often the group of interest or some numerical value but it can also be a matrix with a lot of different targets of interest. In anycase, we can use the y argument from umap function to pass the target of interest. For example here we use subclusters as the target. Please be aware that we need to convert categorical variables to factors.
set.seed(123)supervised_score<-umap(data[,1:6],n_neighbors =30,y =data$subcluster,spread =7,min_dist =3)plot_ly(as.data.frame(supervised_score), x =~V1, y =~V2, color =~data$main_cluster,symbol =~data$subcluster,colors =c("red", "green", "blue", "purple"),type ="scatter", mode ="markers",size =5) %>%layout(title ="",showlegend =FALSE )
Here i had to spread the points a bit so we can see the results a bit better. Compared to the results we saw before, this looks a bit better at least some of the sub clusters (shapes) they moved closer to each other which basically reflects their relationship that we wanted to capture using y. Given some test data we could perform the projection of the test datapoints onto this space and use some form of similarity to perform prediction but we are going to leave this to you to do :)
3.5 Should i trust UMAP?
While UMAP is a powerful tool that preserves both local and global structures in your data, it’s important to approach the results with caution. UMAP does not guarantee perfect global preservation, and it may sometimes produce artifacts, such as random noise clusters, that do not necessarily reflect meaningful patterns in the data. These clusters can arise due to random noise or overfitting during the optimization process, especially when tuning the parameters too aggressively.
UMAP is primarily designed to preserve local relationships, meaning that points close to each other in the high-dimensional space should remain close in the low-dimensional embedding. However, global structure may be distorted, and distant relationships may not always be accurately represented. This is particularly true when using smaller values for the n_neighbors parameter, which can make UMAP focus too much on local clusters, potentially missing broader trends.
It’s also important to avoid tuning UMAP’s parameters to match a desired outcome or preconceived conclusion. Over-tuning the algorithm, especially by manipulating n_neighbors or min_dist to achieve specific patterns, can lead to misleading visualizations that don’t reflect the true structure of the data. Instead, parameter tuning should be guided by the nature of the data and the goal of the analysis, not by an attempt to force specific patterns or clusters.
Lastly, similary to t-SNE avoid doing distance based clustering methods on UMAP. Density based clustering might work but there is no guarantee for that!
Code
library(Rtsne)library(uwot)cat_dt<-read.table("https://raw.githubusercontent.com/PayamEmami/pca_basics/refs/heads/master/data/cat.tsv")set.seed(123)tsne_results_cat <-Rtsne(as.matrix(cat_dt),perplexity =30,)set.seed(123)umap_results_cat <-umap(as.matrix(cat_dt),n_neighbors =30,)pca<-prcomp(cat_dt)$x[,1:2]pca_plot <-plot_ly(as.data.frame(pca), x =~PC1, y =~PC2,type ="scatter", mode ="markers",size =5) %>%layout(title ="" )tsne_plot <-plot_ly(as.data.frame(tsne_results_cat$Y), x =~V1, y =~V2, type ="scatter", mode ="markers",size =5) %>%layout(title ="" )umap_plot <-plot_ly(as.data.frame(umap_results_cat), x =~V1, y =~V2, type ="scatter", mode ="markers",size =5) %>%layout(title ="" )colnames(cat_dt)<-c("V1","V2","V3")original_data <-plot_ly(as.data.frame(cat_dt), x =~V1, y =~V2,z=~V3,type ="scatter3d", mode ="markers",size =5) %>%layout(title ="Original data" )original_data
Code
subplot(pca_plot%>%layout(showlegend =FALSE), tsne_plot%>%layout(showlegend =FALSE), umap_plot%>%layout(showlegend =FALSE),titleX = T,titleY = T,margin =0.05)%>%layout(annotations =list(list(x =0.13 , y =1.035, text ="PCA", showarrow = F, xref='paper', yref='paper'),list(x =0.53 , y =1.035, text ="t-SNE", showarrow = F, xref='paper', yref='paper'),list(x =0.85 , y =1.035, text ="UMAP", showarrow = F, xref='paper', yref='paper')))
3.6 Math
Similar to t-SNE, UMAP can also be split into three stages: building a notion of distances both in higher and lower dimension and optimizing the low-dimensional embedding to preserve both local and global structures.
The biggest difference in my opinion is that UMAP is based on much stronger algebraic and topological foundations, using concepts from category theory and simplicial sets to create a more flexible representation of local and global relationships, whereas t-SNE relies primarily on probability distributions to measure similarity between points.
We have actually introduce much of the math in the t-SNE section. So here i’m going to give an overview of what is going on in UMAP.
3.6.1 Manifold of the data
One thing to note is that we believe our data is sitting on a lower-dimensional manifold embedded within a higher-dimensional space. A manifold is a mathematical structure that, at every small enough region, looks like a flat space (Euclidean space). In simpler terms, even though the data may exist in a high-dimensional space, it behaves like a simpler, lower-dimensional shape when viewed locally. For example, the surface of a sphere (like the Earth) is a 2D manifold, because if you zoom in enough on any part of it, it looks flat like a piece of paper, even though globally it’s curved. In the context of our data, this means that while the data might appear high-dimensional and complex, its structure can be understood through a simpler geometric object. This manifold provides a more compact and interpretable representation, capturing both the local details (where the data behaves like flat space) and the global shape (which can have curves and twists). So to sum it up, the manifold is an overall shape of our data. We don’t know this manifold but we are trying to figure out how it looks. For example look at the following curve which visualize a circadian-like trend:
Code
# Load necessary librarieslibrary(plotly)# Function to generate points on a circadian-like curvegenerate_points_circadian <-function(n_dense, n_sparse) {# Dense region (more points) t_dense <-seq(0, 1* pi, length.out = n_dense)# Sparse region (fewer points, covering a similar interval) t_sparse <-seq(1* pi, 2* pi, length.out = n_sparse)# Generate the circadian curve (oscillating sine-cosine) x_dense <- t_dense y_dense <-sin(t_dense) *cos(t_dense /2) x_sparse <- t_sparse y_sparse <-sin(t_sparse) *cos(t_sparse /2)# Combine the dense and sparse points x <-c(x_dense, x_sparse) y <-c(y_dense, y_sparse)# Normalize x and y values to the same scale x <- (x -min(x)) / (max(x) -min(x)) # Normalize x to [0, 1] y <- (y -min(y)) / (max(y) -min(y)) # Normalize y to [0, 1]# Create a data frame with the results data <-data.frame(x = x, y = y, density =c(rep("Dense", n_dense), rep("Sparse", n_sparse)))return(data)}# Function to generate the entire manifold (for plotting the line)generate_manifold_curve <-function(n_points) { t <-seq(0, 2* pi, length.out = n_points) x <- t y <-sin(t) *cos(t /2)# Normalize x and y values to the same scale x <- (x -min(x)) / (max(x) -min(x)) # Normalize x to [0, 1] y <- (y -min(y)) / (max(y) -min(y)) # Normalize y to [0, 1] data <-data.frame(x = x, y = y)return(data)}# Set number of points in dense and sparse regionsn_dense <-100n_sparse <-100# Generate the pointsdata <-generate_points_circadian(n_dense, n_sparse)# Generate the manifold curvemanifold <-generate_manifold_curve(500) # 500 points for a smooth curve# Plot using plotlyp <-plot_ly() %>%# Add the manifold lineadd_trace(data = manifold, x =~x, y =~y, type ='scatter', mode ='lines',line =list(color ='black', width =2), name ='Manifold') %>%# Add the points with varying densityadd_trace(data = data, x =~x, y =~y, type ='scatter', mode ='markers', color =~density, colors =c('blue', 'red'),marker =list(size =10, opacity =0.7), name ='Points') %>%# Layoutlayout(title ="",xaxis =list(title ="X"),yaxis =list(title ="Y"),showlegend =FALSE)# Show the plotp
The idea here is that we are interested in characterizing the underlying structure of the data, which in this case follows a periodic, wavelike pattern. In this context, we can interpret the curve as lying on a manifold. A manifold is a special type of topological space. A topological space is an abstract way of talking about the “shape” of a set of points. If we have a set \(X\), which is just a collection of points (for example, the real numbers, or points on a piece of paper), in order to study this space, we need a way to talk about which points are “close” to each other. Instead of using specific distances, we use something called open sets. An open set is a special kind of subset of \(X\). Think of it like a region around a point where everything inside the region is considered “close” to that point, but the edges of the region are not included. For a set of points to be considered “open” in a topological space, it has to follow these three rules: 1. The whole space \(X\) and the empty set \(\emptyset\) must be open. This just means that there’s a way to think of everything and nothing as being “open” in a sense. 2. If you take any number of open sets and combine them, the result should still be open. This allows us to talk about combining neighborhoods around points. 3. If you take a finite number of open sets and look at their overlap (where they share points), this overlap must also be an open set.
An example of an open set in the context of topology and metric spaces is called open ball. In a metric space, an open ball is defined as the set of all points that are within a certain distance (radius) from a given point (the center of the ball). Formally, if we have a metric space \((X, d)\), where \(d\) is the distance function (or metric), the open ball centered at a point \((x \in X)\) with radius \(r > 0\) is defined as:
\[
B(x, r) = \{ y \in X \mid d(x, y) < r \}
\]
This set contains all the points \(y\) whose distance from \(x\) is strictly less than \(r\). This basically satisfies the definition of an open set: For every point \(p\) inside the open ball \(B(x, r)\), we can find a smaller open ball around \(p\) that is still entirely contained within \(B(x, r)\).
Here is an example of open ball for our data:
Code
chosen_point <- data[50, ] ball_radius <-0.05# Set the radius of the open ball# Create the open ball as a circle around the chosen pointtheta <-seq(0, 2* pi, length.out =100)circle_x <- chosen_point$x + ball_radius *cos(theta)circle_y <- chosen_point$y + ball_radius *sin(theta)# Plot using plotlyp <-plot_ly() %>%# Add the manifold lineadd_trace(data = manifold, x =~x, y =~y, type ='scatter', mode ='lines',line =list(color ='black', width =2), name ='Manifold') %>%# Add the points with varying densityadd_trace(data = data, x =~x, y =~y, type ='scatter', mode ='markers', color =~density, colors =c('blue', 'red'),marker =list(size =10, opacity =0.7), name ='Points') %>%# Add the open ball (circle) around the chosen pointadd_trace(x = circle_x, y = circle_y, type ='scatter', mode ='lines',line =list(color ='green', dash ='dash', width =2), name ='Open Ball') %>%# Highlight the chosen pointadd_trace(x =~chosen_point$x, y =~chosen_point$y, type ='scatter', mode ='markers',marker =list(size =15, color ='green', symbol ='circle'), name ='Chosen Point') %>%# Layout with equal axis scalinglayout(title ="",xaxis =list(title ="X", scaleanchor ="y"), # Link x and y axes for equal scalingyaxis =list(title ="Y", scaleratio =1),showlegend =FALSE) # Enforce equal aspect ratio for x and y axes# Show the plotp
Once we have open sets or in this case open balls, we can use them to study a topological space. One way to do this is by breaking the space into smaller, manageable pieces using open covers. An open cover is a collection of open sets that completely covers the entire space. The idea behind open covers is that we can study complex spaces by looking at how these smaller open sets relate to each other. Formally an open cover of a topological space \(X\) is a collection of open sets \(\{ U_i \}_{i \in I}\) such that: \[
X = \bigcup_{i \in I} U_i
\] Every point in \(X\) is contained in at least one open set \(U_i\).
We can see an example of cover here:
Code
chosen_indices <-seq(1,200,by=2) # Choose several points for the open coversball_radius <-0.05# Set the radius of the open covers# Initialize the plotp <-plot_ly() %>%# Add the manifold lineadd_trace(data = manifold, x =~x, y =~y, type ='scatter', mode ='lines',line =list(color ='black', width =2), name ='Manifold') %>%# Add the points with varying densityadd_trace(data = data, x =~x, y =~y, type ='scatter', mode ='markers', color =~density, colors =c('blue', 'red'),marker =list(size =10, opacity =0.7), name ='Points')# Add the open covers (circles) around the chosen pointsfor (index in chosen_indices) { chosen_point <- data[index, ] theta <-seq(0, 2* pi, length.out =100) circle_x <- chosen_point$x + ball_radius *cos(theta) circle_y <- chosen_point$y + ball_radius *sin(theta)# Add each open cover to the plot p <- p %>%add_trace(x = circle_x, y = circle_y, type ='scatter', mode ='lines',line =list(color ='green', dash ='dash', width =2), name ='Open Cover')}# Highlight the chosen points for the open coversp <- p %>%add_trace(x =~data[chosen_indices, ]$x, y =~data[chosen_indices, ]$y, type ='scatter', mode ='markers',marker =list(size =15, color ='green', symbol ='circle'), name ='Cover Centers')# Layout with equal axis scalingp <- p %>%layout(title ="",xaxis =list(title ="X", scaleanchor ="y"), # Link x and y axes for equal scalingyaxis =list(title ="Y", scaleratio =1),showlegend =FALSE) # Enforce equal aspect ratio for x and y axes# Show the plotp
When we look at an open cover, one important thing to study is how the open sets overlap. The nerve of an open cover is a mathematical structure that tells us about these overlaps. Nerve Theorem says that If \(\mathcal{U}\) is an open cover of a good topological space (like a paracompact space), and all finite intersections of open sets in \(\mathcal{U}\) are contractible or empty, then the nerve \(\mathcal{N(U)}\) has the same homotopy type (two spaces have the same homotopy type if they can be continuously deformed into each other. They share the same “shape” in a topological sense) as \(X\). This simply means that if the open sets in the cover overlap in a “nice” way, then the nerve of the cover has the same essential shape as the original topological space.
So how do we make nerves?! The nerve \(\mathcal{N(U)}\) of an open cover \(\mathcal{U} = \{ U_i \}\) is an abstract simplicial complex constructed as follows:
Vertices: Each open set \(U_i\) corresponds to a vertex \(v_i\).
Simplices: A finite subset \(\{ v_{i_0}, v_{i_1}, \dots, v_{i_k} \}\) spans a \(k\)-simplex if the intersection of the corresponding open sets is non-empty:
This nerve records the way open sets overlap, translating continuous topological information into a discrete combinatorial object.
The fact that a nerve is simplicial complex brings us to the main concept behind UMAP and that is simplicial complexes. A simplicial complex \(K\) is a collection of simplices (points, line segments, triangles, and their higher-dimensional counterparts).
We can think about the simplices as strcutures that connect the data points together:
0-simplex: A point.
1-simplex: A line segment between two points.
2-simplex: A filled triangle formed by three points.
3-simplex: A filled tetrahedron formed by four points.
k-simplex: The convex hull of \(k+1\) affinely independent points.
The geometric realization of a simplicial complex is a topological space formed by “gluing together” the simplices in a way that matches their combinatorial connections. We are going to get back to this later
Here we are not going to create a nerve in classical sense but rather we want to follow its steps conceptually. We want to create a cover through open sets and then fix the interaction (overlap) between these sets. Let’s go step by step:
3.6.2 Defining the Cover (Open Balls/Open sets)
The first and probably the most important thing here is to create open sets. If you remember, we need to create these sets whose cover is going to involve the entire space. In the best case scenario we want to have infinite data points to be uniformly distributed over the manifold. In this case, we just create open balls with some radios around each point and anything within would be included in a set. This is visualized in the previous plot. In reality however, we are dealing with finite number of data points which are non-uniformly distributed on the manifold:
Code
# Load necessary librarieslibrary(plotly)# Function to generate points on a circadian-like curvegenerate_points_circadian <-function(n_dense, n_sparse) {# Dense region (more points) t_dense <-seq(0, 1* pi, length.out = n_dense)# Sparse region (fewer points, covering a similar interval) t_sparse <-seq(1* pi, 2* pi, length.out = n_sparse)# Generate the circadian curve (oscillating sine-cosine) x_dense <- t_dense y_dense <-sin(t_dense) *cos(t_dense /2) x_sparse <- t_sparse y_sparse <-sin(t_sparse) *cos(t_sparse /2)# Combine the dense and sparse points x <-c(x_dense, x_sparse) y <-c(y_dense, y_sparse)# Normalize x and y values to the same scale x <- (x -min(x)) / (max(x) -min(x)) # Normalize x to [0, 1] y <- (y -min(y)) / (max(y) -min(y)) # Normalize y to [0, 1]# Create a data frame with the results data <-data.frame(x = x, y = y, density =c(rep("Dense", n_dense), rep("Sparse", n_sparse)))return(data)}# Function to generate the entire manifold (for plotting the line)generate_manifold_curve <-function(n_points) { t <-seq(0, 2* pi, length.out = n_points) x <- t y <-sin(t) *cos(t /2)# Normalize x and y values to the same scale x <- (x -min(x)) / (max(x) -min(x)) # Normalize x to [0, 1] y <- (y -min(y)) / (max(y) -min(y)) # Normalize y to [0, 1] data <-data.frame(x = x, y = y)return(data)}# Set number of points in dense and sparse regionsn_dense <-100n_sparse <-20# Generate the pointsdata <-generate_points_circadian(n_dense, n_sparse)# Generate the manifold curvemanifold <-generate_manifold_curve(500) # 500 points for a smooth curve# Plot using plotlyp <-plot_ly() %>%# Add the manifold lineadd_trace(data = manifold, x =~x, y =~y, type ='scatter', mode ='lines',line =list(color ='black', width =2), name ='Manifold') %>%# Add the points with varying densityadd_trace(data = data, x =~x, y =~y, type ='scatter', mode ='markers', color =~density, colors =c('blue', 'red'),marker =list(size =10, opacity =0.7), name ='Points') %>%# Layoutlayout(title ="",xaxis =list(title ="X"),yaxis =list(title ="Y"),showlegend =FALSE)# Show the plotp
Here the points are often not distributed evenly across the input space. Instead, the data has regions of high density (where many points are close together) and regions of low density (where points are sparse and far apart). If we use a fixed-radius open ball for neighborhood construction (e.g., \(r\)-balls in a metric space), it doesn’t adapt well to non-uniform data. In dense regions, the fixed radius might include too many points, creating very large and noisy neighborhoods. In sparse regions, the fixed radius might include too few or even no points, making it hard to capture the structure of the data. So we basically need to change our defintion of open set slightly. This means that instead of having
We want to allow for a gradual decay of membership in the set depending how far is it from the center, meaning that points far from the center of the ball have some small degree of membership in the set. This we are going to denote as a fuzzy open set. This basically means that instead of data points being exactly present or absent in a set, we will have a weight or probability showing how likely it is for a point to belong to that set.
Now, since we are working on a manifold, a space that can be locally approximated by Euclidean space but may have a more complex global structure, we need to adjust our notion of distance to account for the manifold’s curvature and geometry (Riemannian geometry).
On a Riemannian manifold \(M\), each point \(x \in M\) is associated with a local metric (a way of measuring distances) that depends on the point’s position. The distance between two points \(x\) and \(x_0\) is not simply the straight-line (Euclidean) distance as in flat space but is instead the geodesic distance, denoted by \(d_M(x, x_0)\). This geodesic distance measures the shortest path on the manifold between \(x\) and \(x_0\), taking into account any curvature or distortions in the space.
This is an example of how geodesic distance is different from Euclidean:
Code
library(plotly)# Function to generate points for the manifoldgenerate_manifold_surface <-function(n_points) { t <-seq(0, pi, length.out = n_points) x <- t y <-sin(t) z <-0.3*cos(2* t) # Curved shape for manifold data <-data.frame(x = x, y = y, z = z)return(data)}# Function to generate the geodesic curve (curved path)generate_geodesic_curve <-function() { t <-seq(0, pi, length.out =100) x <- t y <-sin(t) z <-0.3*cos(2* t) # Geodesic follows the manifold surface data <-data.frame(x = x, y = y, z = z)return(data)}# Function to generate the Euclidean linegenerate_euclidean_line <-function() { x <-c(0, pi) y <-c(0, sin(pi)) # Points A (0,0) to B (pi, sin(pi)) z <-c(0.3*cos(2*0), 0.3*cos(2* pi)) # Straight line between A and B data <-data.frame(x = x, y = y, z = z)return(data)}# Function to generate start and end points (A and B)generate_points_A_B <-function() { x <-c(0, pi) # Start (A) and end (B) points y <-c(0, sin(pi)) z <-c(0.3*cos(2*0), 0.3*cos(2* pi)) data <-data.frame(x = x, y = y, z = z, label =c("A", "B"))return(data)}# Generate the manifold surface, geodesic curve, Euclidean line, and points A and Bmanifold <-generate_manifold_surface(100)geodesic <-generate_geodesic_curve()euclidean <-generate_euclidean_line()points_AB <-generate_points_A_B()# Plot using plotlyp <-plot_ly() %>%# Add the manifold surfaceadd_trace(data = manifold, x =~x, y =~y, z =~z, type ='scatter3d', mode ='lines',line =list(color ='lightgrey', width =100), name ='Manifold',opacity=0.5) %>%# Add the geodesic curveadd_trace(data = geodesic, x =~x, y =~y, z =~z, type ='scatter3d', mode ='lines',line =list(color ='green', width =5), name ='Geodesic Distance') %>%# Add the Euclidean straight lineadd_trace(data = euclidean, x =~x, y =~y, z =~z, type ='scatter3d', mode ='lines',line =list(color ='blue', dash ='dash', width =4), name ='Euclidean Distance') %>%# Add points A and Badd_trace(data = points_AB, x =~x, y =~y, z =~z, type ='scatter3d', mode ='markers+text',marker =list(color ='red', size =8), text =~label, textposition ='top middle',name ='Points A and B') %>%# Layoutlayout(scene =list(xaxis =list(title ="X"),yaxis =list(title ="Y"),zaxis =list(title ="Z")),title ="Geodesic vs Euclidean Distance with Points A and B",showlegend =TRUE)# Show the plotp
So when we want to define a fuzzy open set on a manifold, the distance function \(d_M(x, x_0)\) will be used in the membership function to reflect how distances are measured in the curved space of the manifold. Mathematically, we define a fuzzy open set \(A\) on the manifold \(M\) by specifying a membership function \(\mu_A: M \to [0,1]\), where the degree of membership of a point \(x \in M\) is determined by the geodesic distance \(d_M(x, x_0)\) from a central point \(x_0 \in M\). A common choice for the membership function is an exponential decay based on the geodesic distance:
where: - \(d_M(x, x_0)\) is the geodesic distance between \(x\) and the center \(x_0\) of the fuzzy set. - \(\alpha > 0\) is a constant controlling the rate of decay of membership.
if we take \(\alpha\) as a fraction then this formula looks quite similar to that of t-SNE:
So similar to t-SNE this formula describes how the membership of a point decays smoothly as its geodesic distance from the center \(x_0\) increases. The key difference from the Euclidean case is that the distance \(d_M(x, x_0)\) now respects the curvature of the manifold.
One of the cool features of Riemannian geometry is that the manifold behaves locally like Euclidean space (zoom in on the manifold to see). In a small neighborhood around \(x_0\), the geodesic distance \(d_M(x, x_0)\) closely approximates the Euclidean distance. This allows us to use the same intuition as in Euclidean space for points near \(x_0\), where membership decays smoothly. However, as we move farther from \(x_0\), the global structure of the manifold can cause the geodesic distance to behave quite differently from the Euclidean distance. This basically means that on a curved manifold like the plot above, the geodesic distance between two points wraps around the curve, and the decay in membership reflects this curvature. On more complex manifolds, the distance might vary in ways that reflect the manifold’s underlying structure, and the fuzzy membership decays accordingly.
But assuming that we want to focus on local structure (neighborhood), we can change this formula to reseble that of t-SNE just for the sake of making more clear:
Here i just replaced \(\alpha\) with \(\sigma\) and plugged in the equation for Euclidean distance. This gives us a formula similar to the Gaussian kernel, which essentially provides the probability that a point \(x\) belongs to the fuzzy set \(A\). In other words, the function assigns higher probabilities to points closer to \(x_0\), and lower probabilities to those farther away, with the parameter \(\sigma\) controlling the spread of these probabilities.
We can write this in a more general way let’s say that for point \(x_i\) we want create an open ball, to calculate the probability of \(x_j\) being part of the open ball \(i\) we can write
\[
w(x_j,x_{ij}) = \exp\left(-\frac{d(x_j,x_{ij})}{\sigma}\right)
\] where \(d(x_j,x_{ij})\) is some function measuring distance between \(x_i\) and \(x_j\).
So far so good, we have a measure that works on local neighborhoods which more or less follows Riemannian metric in local parts of the manifold. Now that we are working on a manifold, we have to make sure that we satifiy its topological properties. We already talked about that a manifold must resemble Euclidean space \(\mathbb{R}^n\) in small neighborhoods around each point. We however, have to make sure that within any small neighborhood of a point on the manifold, we can draw continuous paths between any two points within that neighborhood. That means that if we zoom in on a super small local neighborhood (like a single point), there is always a neighboring point close enough that we can put them part of a set. This in turn means that we for \(x_i\) we believe is that there is always a point close enough with any distance \(\rho_i\) such that there is 100% probability that they are part of the same set. Therefore just to make sure that we satisfy this property, we are going to adjust the probablity formula above so that the closet neighbro will always get a probablity of 1.
In this formula, as before \(d(x_j, x_{ij})\) represents the distance between point \(x_j\) and its neighbor \(x_{ij}\). The term \(\rho_i\) is the distance to the nearest neighbor of \(x_j\), ensuring that the closest neighbor always has a maximum probability of 1. By subtracting \(\rho_i\) from the distance, we ensure that any distance less than or equal to \(\rho_i\) results in a probability of 1, as \(\max(0, d(x_j, x_{ij}) - \rho_i)\) will be zero for the closest neighbors.
As said before \(\sigma_i\) parameter controls the spread or sharpness of the decay of the probability for neighbors that are further away. As the distance between \(x_j\) and \(x_{ij}\) increases, the probability decreases exponentially, following the Riemannian structure of the local neighborhood. This adjustment guarantees that the local-connectivity property is satisfied, ensuring that the nearest neighbors remain tightly connected and thus preserving the manifold’s local geometry while allowing the global structure to be represented appropriately. Therefore, specially in very high dimensional data where points tend to become isolated, we measure that that they are at least connect to one neighbor.
With this adjustment comes a really cool effect: normalizing distances in sparse regions of the manifold where data points are very far from each other. In that case, everything will be measured relative to the nearest neighbor (which can local very far also), ensuring that even when points are sparsely distributed, the structure remains consistent. This normalization prevents distances from blowing up in sparse areas, which would otherwise distort the local geometry.
The above equation is exactly what UMAP uses to measure the distances between the data points. We are however not done here is another property we need to take care of. That is to set \(\sigma_i\).
If we pause for a second and think about what we have done now, we wanted to create open sets meaning that we need to focus on small little region around each point. We can select \(k\) closest points to the current one and then calculate \(\sigma\) and assign the probability based on these \(k\) distances and then create our open set based on these distances. If you remember, we said this topological approach would work if data is uniformly distributed on the manifold. This basically means that we expect that for each data point, neighbors affect it similarly on average.
Mathematically, we are dealing with a finite set of distances, we can simply sum the probabilities (weights) for each set and compare the sums directly.
For set \(i\), the total probability (sum of weights) can be written as:
Here, \(n_i\) and \(n_k\) are the number of neighbors considered in sets \(i\) and \(l\), respectively.
If the data was uniformly distributed we would expect
\[
S_i=S_l=C
\]
That is the expectation of weights in each set would be equal to the same constant. This constant is global and does not change no matter where on the manifold we build our sets.
If all the neighbors contribute equally to \(S_i\), the most straightforward interpretation would imply that the distances \(d(x_j, x_{ij})\) are relatively similar for all \(j\), meaning that \(d(x_j, x_{ij}) \approx \rho_i\). As a result: \[
S_i = \sum_{j=1}^{n_i} 1 = n_i.
\] Each term contributes equally and fully, leading to the sum being exactly equal to \(n_i\), the number of neighbors.
So does this mean that we should put the value equal to the number of neighbors \(k\)? It turned out that this is a bad idea. If we think about it for a second, if we use \(k\), because in dense regions there are a lot of neighboring points, we tend to quickly pick a few and get to the \(k\), meaning that the \(\sigma\) will be relatively small. In sparse regions, however, we need to expand the neighborhood much further to reach \(k\) neighbors, resulting in a much larger \(\sigma\). This creates an imbalance: in dense regions, small \(\sigma\) values cause the model to overemphasize local structure, while in sparse regions, large \(\sigma\) values cause distant points to exert too much influence, leading to distortions.
To address this, we use \(\log_2(k)\) instead. The logarithmic scaling grows much more slowly than linear scaling, which moderates the influence of neighbors. For example, if \(k = 4\), \(\log_2(4) = 2\), but if \(k = 16\), \(\log_2(16) = 4\). Notice how \(\log_2(k)\) grows much slower as \(k\) increases. This ensures that in dense regions, the sum of influences doesn’t explode, and in sparse regions, neighbors still contribute meaningfully without overwhelming the embedding. This smoother scaling prevents dense regions from becoming too tightly packed and allows sparse regions to remain connected, maintaining a balanced and meaningful representation of both local and global structure in the embedding.
So to summarize this section, we are going to create fuzzy open sets, with one set for each data point. The membership strength is going to be measured by
where \(d(x_j, x_{ij})\) is the distance between the data point \(x_j\) and its \(i\)-th neighbor up to \(k\) neighbors, and \(\rho_i\) represents the distance to the nearest neighbor. The parameter \(\sigma_i\) controls the spread of the membership function. We will adjust \(\sigma_i\) such that the sum of the weights across the neighborhood equals \(\log_2(k)\), where \(k\) is the number of neighbors, ensuring that each point’s neighborhood contributes smoothly and consistently to the embedding.
This wraps up our section on creating fuzzy open sets. Let’s try to implement this:
# Load required packageslibrary(FNN) # For exact k-nearest neighborslibrary(matrixStats) # For row operations# Step 1: Find k-nearest neighbors (KNN) and distances using exact methodget_knn_exact <-function(X, k) { knn_result <-get.knn(X, k = k, algorithm ="brute")list(knn = knn_result$nn.index, dists = knn_result$nn.dist)}# Step 2: Perform binary search to find sigma such that sum(exp(...)) = log2(k)find_sigma <-function(dists, rho, k, tolerance =1e-5, max_iter =1000) { log2_k <-log2(k) low <-0 high <-1000 sigma <-1 target <- log2_kfor (i in1:max_iter) { psum <-sum(exp(-(pmax(0, dists - rho)) / sigma))if (abs(psum - target) < tolerance) {break }if (psum > target) { high <- sigma sigma <- (low + high) /2 } else { low <- sigmaif (high ==1000) { sigma <- sigma *2 } else { sigma <- (low + high) /2 } } }return(sigma)}# Step 3: Calculate fuzzy membership strengthcalculate_membership_strength <-function(dists, sigmas, rhos) {exp(-(pmax(0, dists - rhos)) / sigmas)}# Main function to compute fuzzy open sets and adjust sigmafuzzy_open_sets <-function(X, k) { knn_data <-get_knn_exact(X, k) knn_dists <- knn_data$dists knn <- knn_data$knn n <-nrow(knn_dists) sigmas <-numeric(n) rhos <-rowMins(knn_dists) # The nearest neighbor distance# Adjust sigma for each pointfor (i in1:n) { sigmas[i] <-find_sigma(knn_dists[i, ], rhos[i], k) }# Compute membership strength memberships <-matrix(0, n, n)for (i in1:n) { memberships[i, knn[i, ]] <-calculate_membership_strength(knn_dists[i, ], sigmas[i], rhos[i]) }return(list(memberships = memberships, sigmas = sigmas, rhos = rhos))}k <-5# Number of neighborsresult <-fuzzy_open_sets(data[,1:2], k)# Output the results#print(result$memberships) # Fuzzy membership matrix#print(result$sigmas) # Sigma values#print(result$rhos) # Rho values (distance to nearest neighbor)
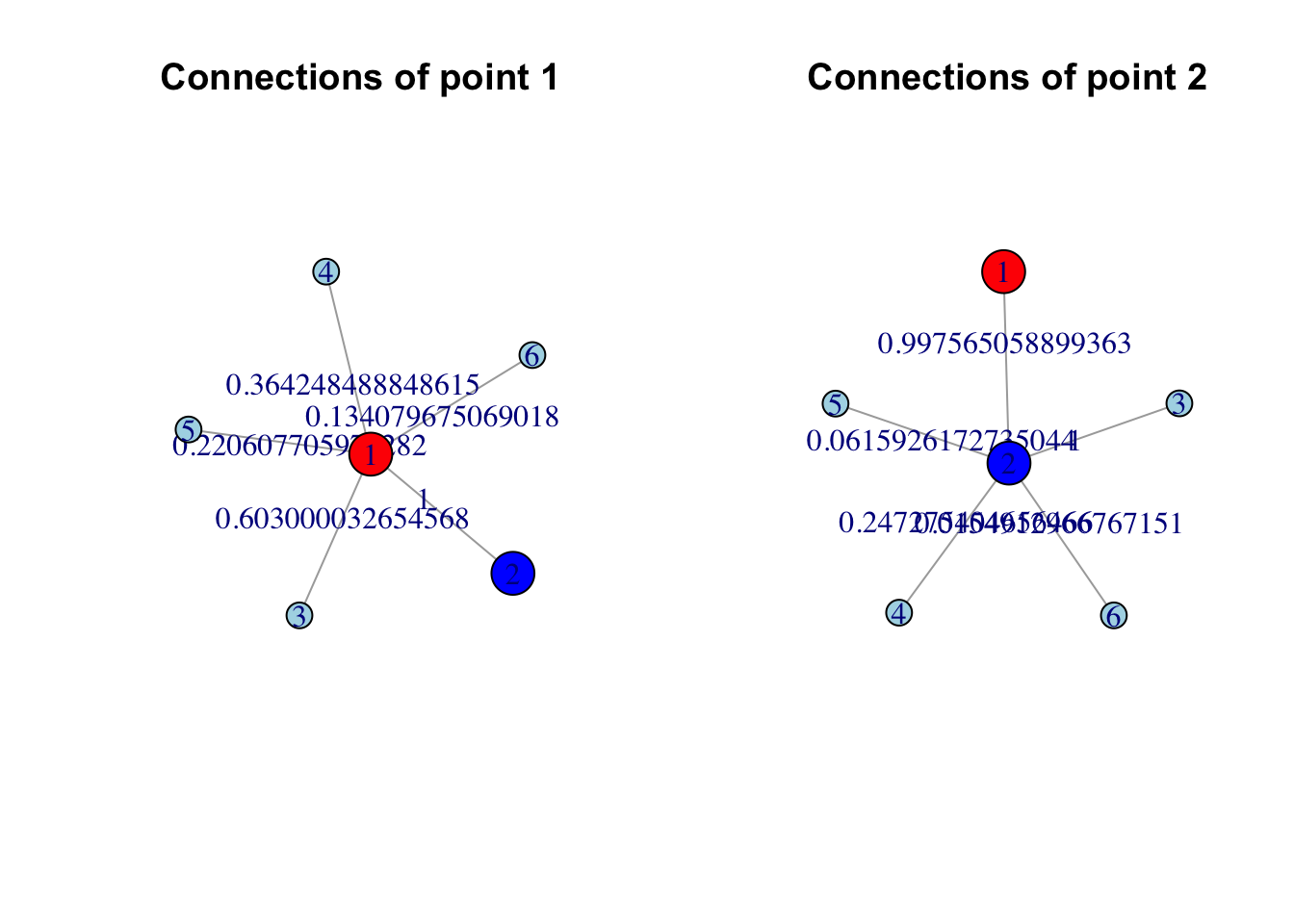
This effectively gives us multiple sets, one for each data points. for example if we set \(k=5\) for data point 1 we have set a set of data points [2, 3, 4, 5, 6] and each of these data points have a membership probability of being part of this set ([2: 1, 3: 0.603, 4: 0.364, 5: 0.221, 6: 0.134]). Similarly we have a set for data point 2 we have [1: 0.998, 3: 1, 4: 0.247, 5: 0.062, 6: 0.015]. Notice that the closes neighbor will always get the probability of \(1\).
At this stage, we begin by considering open sets in a topological sense. In topology, an open set is a collection of points where, for any given point within the set, we can find a surrounding neighborhood that still lies entirely within the set.
Now, let’s think of each point in our data as being surrounded by an open set, and the overlap between these open sets represents some degree of similarity or connection between the points. When two points have significant overlap in their open sets, they are likely to be strongly connected, and when the overlap is small, the connection is weaker.
To represent these overlapping open sets in a more structured way, we use simplicial sets. A simplicial set is a mathematical structure made up of points (called vertices) and their connections (called simplices) in different dimensions.
A 0-simplex represents a single point, or vertex, in our simplicial set. It’s the most basic building block. In our case, the 0-simplices correspond to the individual points in the data. We aren’t yet considering any relationships between the points but just the points themselves. Each point can be thought of as its own 0-simplex. A 1-simplex represents a connection between two 0-simplices, or an edge between two points. In the context of fuzzy simplicial sets, these edges are not just binary (either existing or not); they are weighted to reflect the strength of the connection. If two points have a lot of overlap between their open sets (meaning they are very similar or close to each other), the 1-simplex between them will have a higher weight. Conversely, if the overlap is smaller, the weight of the 1-simplex will be lower.
These connections between points can be naturally represented as graphs. In graph theory each point (or vertex) in our set is represented as a node in the graph (this corresponds to our 0-simplices). The relationships or connections between the points (1-simplices) are represented as edges in the graph.
To make things more concrete, let’s focus on two sets of points. For each set, we can represent the points as 0-simplices and their connections as 1-simplices.
Code
# Load necessary packagelibrary(igraph)# Assuming `result$memberships` is a matrix where rows are vertices and columns are connections# Connections and weights for vertex 1connections_v1 <-which(result$memberships[1,] !=0)weights_v1 <- (result$memberships[1, connections_v1])# Connections and weights for vertex 2connections_v2 <-which(result$memberships[2,] !=0)weights_v2 <- (result$memberships[2, connections_v2])# Create edge lists for vertex 1 and vertex 2edges_v1 <-c(rbind(rep(1, length(connections_v1)), connections_v1))edges_v2 <-c(rbind(rep(2, length(connections_v2)), connections_v2))# Create the graphs for vertex 1 and vertex 2g_v1 <-graph(edges = edges_v1, directed =FALSE)g_v2 <-graph(edges = edges_v2, directed =FALSE)# Set edge weights as attributesE(g_v1)$weight <- weights_v1E(g_v2)$weight <- weights_v2vertex_colors_v1<-rep("lightblue",length(V(g_v1)))vertex_colors_v1[V(g_v1)==1] <-"red"vertex_colors_v1[V(g_v1)==2] <-"blue"vertex_colors_v2<-rep("lightblue",length(V(g_v1)))vertex_colors_v2[V(g_v2)==1] <-"red"vertex_colors_v2[V(g_v2)==2] <-"blue"vertex_sizes_v1 <-ifelse(V(g_v1) %in%c(1, 2), 25, 15) # Larger for vertex 1 and 2vertex_sizes_v2 <-ifelse(V(g_v2) %in%c(1, 2), 25, 15) # Larger for vertex 1 and 2# Set up two-panel plottingpar(mfrow =c(1, 2))# Plotlayout_v1 <-layout_with_dh(g_v1)plot(g_v1, layout = layout_v1, # Use a stable layoutedge.label =E(g_v1)$weight, # Display weights on edgesvertex.label =V(g_v1), # Show vertex numbersedge.width =E(g_v1)$width, # Adjust edge thicknessvertex.color = vertex_colors_v1, # Highlight vertices 1 and 2vertex.size = vertex_sizes_v1, # Increase size of vertices 1 and 2main ="Connections of point 1") # Title for panel 1layout_v2 <-layout_with_dh(g_v2)plot(g_v2, layout = layout_v2, # Use a stable layoutedge.label =E(g_v2)$weight, # Display weights on edgesvertex.label =V(g_v2), # Show vertex numbersedge.width =E(g_v2)$width, # Adjust edge thicknessvertex.color = vertex_colors_v2, # Highlight vertices 1 and 2vertex.size = vertex_sizes_v2, # Increase size of vertices 1 and 2main ="Connections of point 2") # Title for panel 2
In our case, since we are working with fuzzy simplicial sets, the edges in the graph are weighted. This means that the strength of the connection between two points is reflected in the weight of the edge that links them. Points with stronger overlaps in their open sets will have edges with larger weights, while weaker connections will have smaller weights.
We are only showing two opens sets which we built for point 1 and point 2. Each of these simplicial sets capture certain part of the manifold. However they are disconnected to have unified topological representation (a nerve) we need to fix how these two sets are interacting and we have to do that for all the pairs of sets.
3.6.3 Fixing the interaction
As shown in the previous graph, we have a bunch of simplicial sets, and we want to “fix” their integration by determining a single weight between them. If you look closely at the graph for point 1, you will see that there is an edge from point 1 to point 2 with a certain probability. Similarly, in the simplicial set for point 2, there is an edge going from point 2 back to point 1, but this edge has a different probability.
At this stage, we are not concerned about other points; our focus is on the simplicial sets for point 1 and point 2 only. What we are trying to achieve is a single weight that connects the simplicial set for point 1 to the simplicial set for point 2. These weights, in a fuzzy simplicial set, represent the probability of the points being part of the same set or, more concretely, the likelihood of an edge existing between the two points.
Now, given that we have two edges between point 1 and point 2—one going from point 1 to point 2 with some probability, and the other going from point 2 to point 1 with a different probability—we are interested in determining the likelihood of at least one edge existing between these two points.
From probability theory, we know that the probability of at least one event (in this case, an edge existing) is complementary to the probability of no event (no edge existing). Specifically, the probability of having at least one edge between point 1 and point 2 is:
\[
P(\text{at least one edge}) = 1 - P(\text{no edge exists between points 1 and 2})
\]
For simplicity, let’s define the probabilities of the edges as follows: - \(P_1\) is the probability of an edge going from point 1 to point 2. - \(P_2\) is the probability of an edge going from point 2 to point 1.
The probability that no edge exists between point 1 and point 2 is the product of the probabilities that neither edge is present. That is, the probability of no edge from point 1 to point 2 is \(1 - P_1\), and the probability of no edge from point 2 to point 1 is \(1 - P_2\). Therefore, the combined probability of no edges between points 1 and 2 is:
\[
P(\text{no edges}) = (1 - P_1)(1 - P_2)
\]
Thus, the probability that at least one edge exists between point 1 and point 2 is:
\[
P(\text{at least one edge}) = 1 - (1 - P_1)(1 - P_2) = P1+P2-P1P2
\]
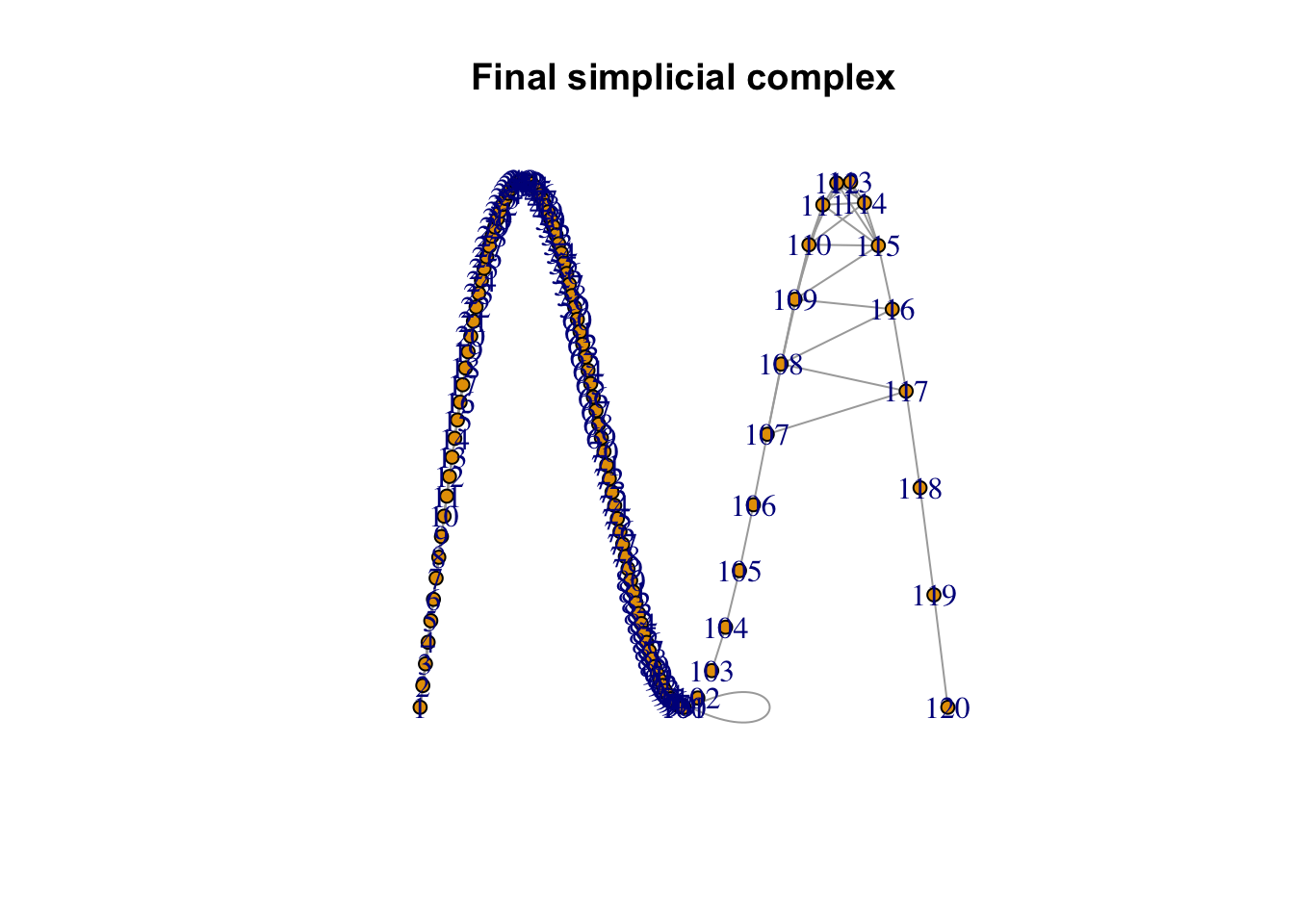
This formula is called probabilistic t-conorm (like OR-like operations but in fuzzy logic) and it gives us a single probability that combines the two directional probabilities into one overall likelihood of there being a connection between the simplicial sets for point 1 and point 2. If we keep can keep doing this for all the pairs of sets we end up with a simplicial complex that according to nerve theory will capture the topology of the data
Code
# Load necessary librarylibrary(igraph)t_conorm_matrix<-result$memberships + result$memberships - (result$memberships*t(result$memberships))t_conorm_matrix[lower.tri(t_conorm_matrix)]<-0# Create an edge list based on non-zero t-conorm valuesedges <-which(t_conorm_matrix !=0, arr.ind =TRUE)edge_weights <- t_conorm_matrix[edges]# Create the graphg <-graph_from_edgelist(as.matrix(edges), directed =FALSE)E(g)$weight <- edge_weights # Assign the t-conorm values as weights# Set node and edge properties for plottingvertex_color <-"lightblue"# Set vertex colorplot(g,layout=as.matrix(data[,1:2]),vertex.label =V(g), # Show vertex numbersvertex.size =5, # Vertex sizemain ="Final simplicial complex") # Title
This process is similar to t-SNE when it make the probabilities symmetric but uses much more statistically valid approach. We are now ready to move to the second part of the algorithm.
3.6.4 Low dimensional similarities
Similar to t-SNE, we need to come up with a function that measures the similarity between points in the low-dimensional space. Let’s start with the following function:
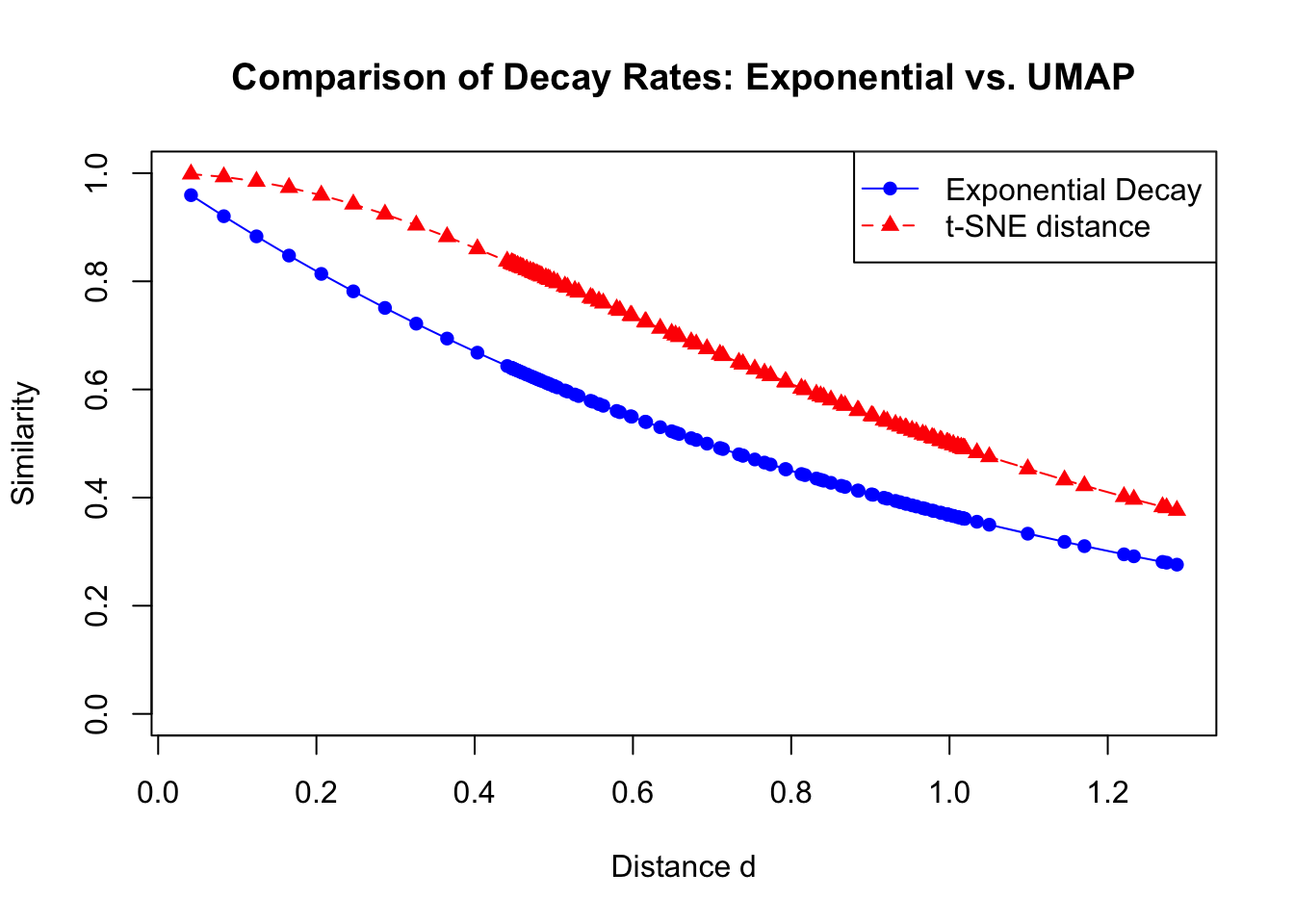
This is a rational function, meaning that it is a ratio of two polynomials. in this case, the numerator is a constant (1), and the denominator is a polynomial of degree 2 in terms of the distance between points \(y_i\) and \(y_j\). Rational functions are benfitial because they are computationally efficient. This means that they involve only basic arithmetic operations, are easy to compute, and their derivatives are straightforward to calculate. This simplicity is beneficial during the optimization process of dimensionality reduction algorithms, where computational efficiency is important.
However, there is a problem with using this function. If you remember, in the high-dimensional space, the similarities between points are defined using an exponential decay function:
\[
p_{ij} = \exp\left(-d \right)
\]
Where \(d\) is a distance. This exponential function decays rapidly with distance, meaning that the similarity between points decreases quickly as the distance increases. The rate of decay of the exponential function is such that distant points have negligible similarity, which helps in preserving the local structure of the data.
In contrast, our initial low-dimensional similarity function $ q_{ij} $ decays polynomially with distance:
\[
q_{ij} = \left(1 + d^2 \right)^{-1}
\]
This function decreases at a slower rate compared to the exponential function. Specifically, as the distance \(d\) becomes large, \(q_{ij}\) behaves like \(\| y_i - y_j \|^{-2}\). The slower decay means that distant points in the low-dimensional space might still have significant similarity values, which can lead to distortions in the embedding. This mismatch in decay rates between $ p_{ij}$ and \(q_{ij}\) can cause the low-dimensional representation to inaccurately reflect the true relationships in the high-dimensional data.
Let’s see how the decay compares between these two function:
Code
# Define the vector of distancesd <-as.matrix(dist(data[,1:2]))[1,-1]# Compute exponential decay valuesexp_decay <-exp(-d)# Compute UMAP similarity valuesumap_similarity <-1/ (1+ d^2)# Plot the exponential decay and UMAP similarityplot(d, exp_decay, type ="o", col ="blue", pch =16, lty =1,ylim =c(0, 1), xlab ="Distance d", ylab ="Similarity",main ="Comparison of Decay Rates: Exponential vs. UMAP")lines(d, umap_similarity, type ="o", col ="red", pch =17, lty =2)legend("topright", legend =c("Exponential Decay", "t-SNE distance"),col =c("blue", "red"), pch =c(16, 17), lty =c(1, 2))
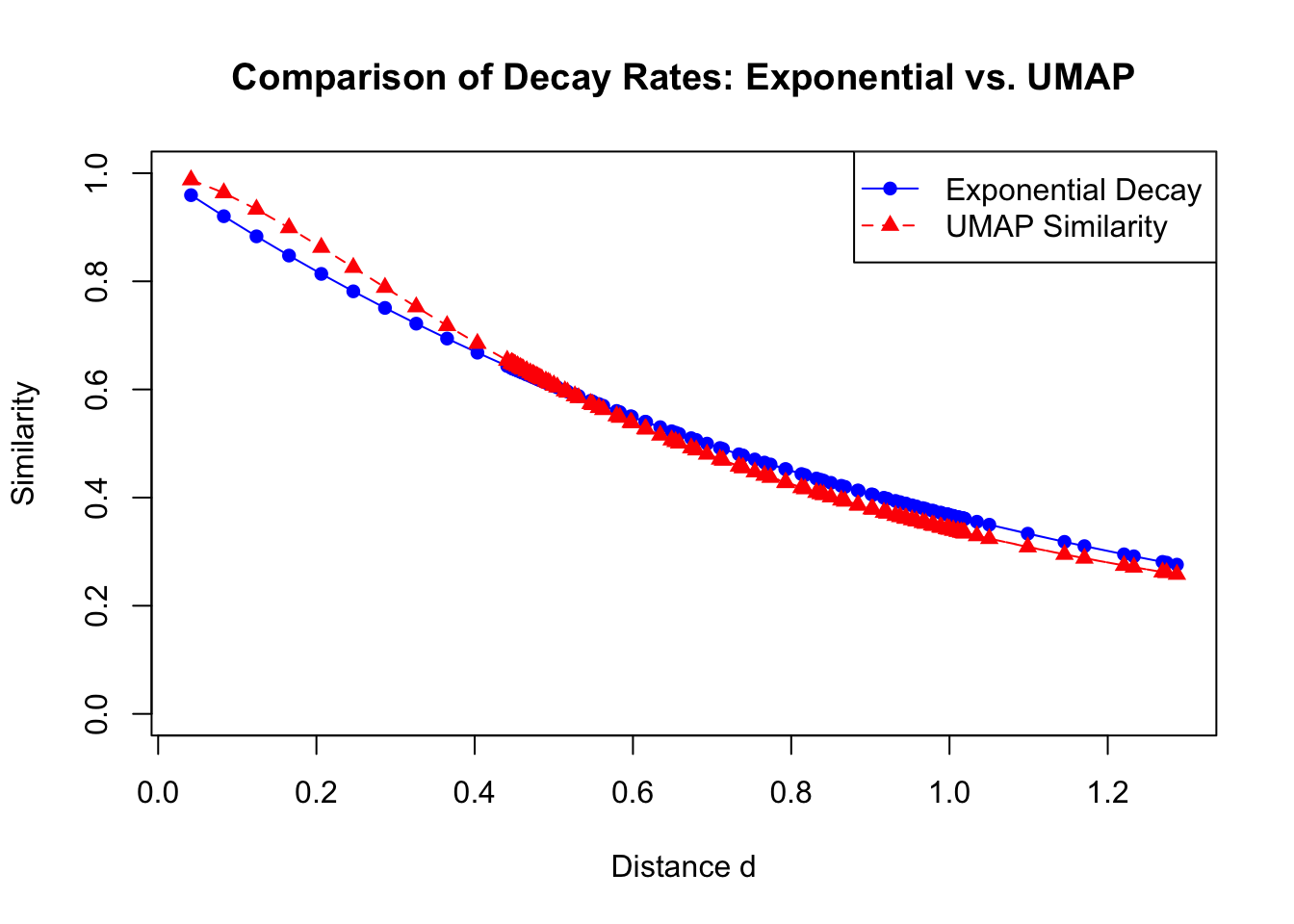
To address this issue, we need to adjust our low-dimensional similarity function to match the rate of decay of the high-dimensional similarities. One way to achieve this is by introducing parameters \(a\) and \(b\) into the function:
By carefully selecting the parameters \(a\) and \(b\), we can control the rate at which \(q_{ij}\) decays with distance. Parameter \(b\) adjusts the exponent of the distance term. By changing \(b\), we can make the decay of \(q_{ij}\) faster or slower. A smaller \(b\) leads to a faster decay, making \(q_{ij]\) decrease more rapidly with distance. Parameter \(a\) scales the distance term. Adjusting \(a\) shifts the rate of decay horizontally, affecting how quickly the similarity decreases as distance increases.
The goal is to find values of \(a\) and \(b\) such that the decay of \(q_{ij}\) approximates the exponential decay of \(p_{ij}\) over the range of distances that are most relevant for preserving the data’s structure. By matching the decay rates, we ensure that the similarities in the low-dimensional space reflect those in the high-dimensional space. This is basically what UMAP uses to measure the similarities. Let’s see an example of this:
Code
# Define the vector of distancesd <-as.matrix(dist(data[,1:2]))[1,-1]# Set parameters for the UMAP functionb <-0.79a <-1.93# Approximate value from UMAP settings# Compute exponential decay valuesexp_decay <-exp(-d)# Compute UMAP similarity valuesumap_similarity <-1/ (1+ a * d^(2* b))# Plot the exponential decay and UMAP similarityplot(d, exp_decay, type ="o", col ="blue", pch =16, lty =1,ylim =c(0, 1), xlab ="Distance d", ylab ="Similarity",main ="Comparison of Decay Rates: Exponential vs. UMAP")lines(d, umap_similarity, type ="o", col ="red", pch =17, lty =2)legend("topright", legend =c("Exponential Decay", "UMAP Similarity"),col =c("blue", "red"), pch =c(16, 17), lty =c(1, 2))
The question is how we are going to set \(a\) and \(b\). Let’s start with our most important assumption that is the data (or distances) are uniformly spread on the manifold. The only thing that we need to set is the spread (remember the spread parameter in UMAP) Let’s generate such data:
spread <-1# Generate a range of distances in low-dimensional spacex <-seq(0, spread *3, length.out =300)hist(x)
We now mimic the UMAP distance function. If you remember: \[
w(x_j, x_{ij}) = \exp\left(-\frac{\max(0, d(x_j, x_{ij}) - \rho_i)}{\sigma_i}\right)
\] As we said in t-SNE section, we don’t need \(\sigma\) in the lower dimension as we assume things to be uniformly distrubted. So we have
\[
w(x_j, x_{ij}) = \exp\left(-{\max(0, d(x_j, x_{ij}) - \rho_i)}\right)
\] If you remember again, any data point with distance lower than \(\rho_i\) is going to get a value of \(w(x_j, x_{ij}) =1\) so we need to set \(\rho_i\). Again because of our assumption, since everything is uniformly distrubted, we can set just one global value for \(\rho_i\), let’s call it min_dist (remember the parameter of UMAP!!). Now we have
Given our distances we can now do the transformation:
min_dist<-0.01y <-numeric(length(x))for (i inseq_along(x)) {y[i] <-exp(-max(0,x[i] - min_dist)) }hist(y)
Now there is only one thing left that is we need to change \(a\) and \(b\) in \(\left(1 + a \| y_i - y_j \|^{2b} \right)^{-1}\) so \(x\) and \(y\) because very similar (UMAP uses non-linear least squere but we are going to just define a cost function sum of suqered and minimize with respect to \(a\) and \(b\).
# Initialize a data frame to store parameter valuesparams_history <-data.frame(a =numeric(), b =numeric())# The low-dimensional distance function dist_low_dim <-function(x, a, b) {1/ (1+ a * x^(2* b)) }# Define the fitting function to minimize fit_function <-function(params) { a <- params[1] b <- params[2] predicted <-dist_low_dim(x, a, b) params_history <<-rbind(params_history, data.frame(a = a, b = b))return(sum((predicted - y)^2)) # Sum of squared errors }# optimize opt_result <-optim(c(1, 1), fit_function)
This optimization tries to make the come up with \(a\) and \(b\) such that the decay looks like an exponential one. You can see the results here:
Code
library(plotly)# Compute the distance vector 'd'd <-as.matrix(dist(data[,1:2]))[1,-1]# Compute the exponential decay reference lineexp_decay <-exp(-d)# Create a data frame to store the framesZ_frames <-data.frame(dim1 =c(), # Placeholder for first dimensiondim2 =c(), # Placeholder for second dimensioniteration =c() # Placeholder for iteration (time step))# Define a grid of 'a' and 'b' values over timea_values <- params_history$ab_values <- params_history$b# Create frames by looping through the a and b valuesfor (i inseq_along(a_values)) { a <- a_values[i] b <- b_values[i]# Compute the UMAP similarity for this iteration sm <-1/ (1+ a * d^(2* b))# Add the computed data for this iteration to the dataframe Z_frames <-rbind(Z_frames, data.frame(dim1 = d, dim2 = sm, iteration = i,col="transfomed d")) Z_frames <-rbind(Z_frames, data.frame(dim1 = d, dim2 =exp(-d), iteration = i,col="exp(-d)"))}# Now create the plotly animation with the exp(-d) linep <-plot_ly(data = Z_frames, x =~dim1, y =~dim2,color=~col,frame =~iteration, # Animate through iterationstype ='scatter', mode ='lines+markers', marker =list(size =10)) %>%layout(title ="UMAP Similarity Animation with exp(-d) Reference",xaxis =list(title ="Distance"),yaxis =list(title ="Transformed distance"),showlegend =TRUE )p
We can create a function so later we can use it in the UMAP implmentation
fit_ab <-function(min_dist, spread) {# The function for piecewise similarity transformation f_transform <-function(x, min_dist) { y <-numeric(length(x))for (i inseq_along(x)) { y[i] <-exp(-max(0,x[i] - min_dist)) }return(y) }# The low-dimensional distance function dist_low_dim <-function(x, a, b) {1/ (1+ a * x^(2* b)) }# Generate a range of distances in low-dimensional space x <-seq(0, spread *3, length.out =300)# Apply the piecewise function to the distances y <-f_transform(x, min_dist)# Define the fitting function to minimize fit_function <-function(params) { a <- params[1] b <- params[2] predicted <-dist_low_dim(x, a, b)return(sum((predicted - y)^2)) # Sum of squared errors }# Optimize the parameters a and b # Starting with initial guesses for a and b opt_result <-optim(c(1, 1), fit_function)# Extract the fitted a and b a <- opt_result$par[1] b <- opt_result$par[2]return(list(a = a, b = b))}
Just to give you a summary, we came up with the function \((1 + a(||y_i - y_j||^2)^b)^{-1}\) that measures the similarity in the lower dimension. We use an optimization algorithm to select the best \(a\) and \(b\). This optimization is irrespective of the data we are working with. It is more capturing the relationship between the polynomial and exponential decay.
We now basically need two more steps to go wrap up UMAP algorithm that is how to optimize lower dimension to look like the high dimension and then where to start optimizing. Let’s start with the optimization.
3.6.5 Optimization (cross-entropy)
Let’s repeated that the primary objective of UMAP is to find a low-dimensional representation of data that preserves the local and global structure present in the high-dimensional space. To achieve this, UMAP constructs a high-dimensional graph representing the data’s structure and then optimizes the low-dimensional embeddings to reflect this structure.
In order to compute how well we are doing in low-dimensional embeddings compared to high-dimensional space, we need a measure. Cross-entropy loss is a measure of how well the low-dimensional embedding represents the high-dimensional data’s relationships. It quantifies the difference between the probabilities of connections (or similarities) in the high-dimensional space and those in the low-dimensional embedding. By minimizing the cross-entropy loss, we ensure that points close together in high-dimensional space remain close in the embedding, and those far apart stay distant.
The cross-entropy loss \(C\) used in UMAP is given by:
\(p_{ij}\) is the probability of points \(i\) and \(j\) being connected in high-dimensional space.
\(q_{ij}\) is the probability in the low-dimensional embedding.
What are attractive and repulsive? The forces that pull similar points closer together in the embedding are called attractive. They ensure that points that are neighbors in the high-dimensional space remain close in the low-dimensional embedding. On the opposite side, repulsive forces push dissimilar points apart in the embedding. They essentially prevent points that are not neighbors from clustering together in the embedding. The idea here is that if we have a lower dimension \(Y\), it should provide a good balance between the attractive and repulsive forces. This means that we are going to start with some random \(Y\) and then update it iteratively based on attractive and repulsive balance so that we can minimize the cross-entropy loss and achieve a meaningful low-dimensional embedding.
Similar to t-SNE To update the low-dimensional embedding \(Y\) we need to calculate the gradient of the cross-entropy loss with respect to \(Y\). The optimization process involves iteratively adjusting \(Y\) using gradient descent, where the attractive and repulsive forces guide the updates.
It turned out that calculating gradients over all pairs \((i, j)\) is computationally expensive for large datasets. We can however try to approximate this using a sampling method. This pretty much means that we select some pairs of points that we think they are neighbours in high dimensional space and optimize the attractive part for them. We then sample some random points (negative samples) and optimize the repulsive part (we believe most pairs in high-dimensional data are dissimilar in high dimensional space). This approach let’s us calculate two gradients one for attractive and one for repulsive pairs.
The attractive Part of the Loss was:
\[
C_{\text{attr}} = -\sum_{(i,j) \in \text{edges}} p_{ij} \log q_{ij}
\]\[
q_{ij}=1 / (1 + a * d^{2 * b})
\] The derivative with respect to \(d_{ij}\) is given by
# Attractive force update for a pair (i, j)update_attractive <-function(y_i, y_j, a, b) { m2ab =-2*a*b codiff <- y_i - y_j codist2 <-sum(codiff*codiff) bm1 = b-1if (codist2 >0) { grad_coeff = (m2ab*codist2^bm1) / ((1+a*(codist2^b)))# Gradient grad <- grad_coeff * codiff } else { grad <-0* codiff # Zero gradient if distance is zero } grad[grad <-4] =-4 grad[grad >4] =4return(grad)}# Repulsive force update for a pair (i, k)update_repulsive <-function(y_i, y_k, a, b, repulsion_strength) { m2ab =-2*a*b codiff <- y_i - y_k codist2 <-sum(codiff*codiff) bm1 = b-1 p2gb<-2*repulsion_strength*bif (codist2 >0) { grad_coeff = p2gb / ((0.001+codist2)*(1+a*(codist2^b)))# Gradient grad <- grad_coeff * codiff } else { grad <-0* codiff }if(grad_coeff>0) { grad[grad <-4] =-4grad[grad >4] =4 }else{ grad<-4 }return(grad)}
Given this now we can go ahead with our sampling-Based Optimization:
Initialize Embeddings: Start with random low-dimensional positions for each data point.
Iterate Over Epochs: Repeat the optimization for a fixed number of epochs.
Attractive Updates:
For each neighbor \(j\) of \(i\), compute the attractive gradient and update embeddings.
Repulsive Updates:
For each point \(i\), sample a few negative samples \(k\) and compute the repulsive gradient.
Code
# Get our high dimensional distances:graph<-result$memberships +t(result$memberships) - (result$memberships*t(result$memberships))N<-nrow(graph)# set a and ba <-1.895526b <-0.8005876# set learning ratealpha <-1# number of componentsn_components <-2# negative_sample_ratenegative_sample_rate <-5# Step 1: Initialize embeddingsset.seed(10)Y <-20*matrix(runif(N * n_components), nrow = N, ncol = n_components)n_epochs <-200# Step 2: Optimization loop (Epochs)# Attractive forcesedges <-which(graph !=0, arr.ind =TRUE) n_edges <-nrow(edges)for (epoch in1:n_epochs) {for (e_idx in (1:n_edges)) { i <- edges[e_idx, 1] j <- edges[e_idx, 2] p_ij_value <- graph[i, j]if(runif(1)<=p_ij_value){# Compute attractive gradient grad_attr <-update_attractive(Y[i, ], Y[j, ], a, b)# Update embeddings Y[i, ] <- Y[i, ] + alpha * grad_attr Y[j, ] <- Y[j, ] - alpha * grad_attr # Symmetric updatefor (n in1:negative_sample_rate) { k <-sample(setdiff(1:N, i), 1) # Ensure k != i grad_rep <-update_repulsive(Y[i, ], Y[k, ], a, b, 1)# Update embedding Y[i, ] <- Y[i, ] + alpha * grad_rep } } } alpha<-1*((1-epoch/n_epochs))}plot(Y)
We can see that the embedding starts having a good shape. There is, however, still room for improvement. While the random initialization provides a starting point, it can sometimes lead to suboptimal positioning of points, especially in capturing the global structure of the data. As optimization progresses, the local relationships between points will likely become more accurate, but achieving a better global alignment might require more iterations. Using a more structured initialization could potentially speed up convergence and provide a more balanced representation of both local and global features from the outset.
3.6.6 Spectral embedding
There are many methods that can be used as initiation of UMAP but spectral embedding is probably one the main choices. The idea is to embed the data into a lower-dimensional space based on the eigenvectors of a graph Laplacian matrix. This matrix encodes the relationships between all data points, offering a globally-aware embedding. We are not going to cover too much of this but to give some intuition about it. Let’s say that \(A\) represents the high-dimensional similarity graph from UMAP. In this case, we want to quickly create a lower dimensonal represetnation of the data using a cost function that shows how well the low-dimensional embedding preserves the structure of the high-dimensional data based on the similarities encoded in \(A\).
Where: - \(A_{ij}\) represents the high-dimensional similarity between points \(i\) and \(j\), as computed by UMAP. - \(\mathbf{x}_i\) and \(\mathbf{x}_j\) are the coordinates of points \(i\) and \(j\) in the low-dimensional embedding. - \(|| \mathbf{x}_i - \mathbf{x}_j ||^2\) is the squared Euclidean distance between these points in the low-dimensional space.
The cost function is a penalty for how much the low-dimensional embedding \(\mathbf{X}\) distorts the high-dimensional relationships encoded in \(A\). If two points \(i\) and \(j\) are highly similar in the high-dimensional space (i.e., \(A_{ij}\) is large), the cost function wants these points to be close together in the low-dimensional space (i.e., \(|| \mathbf{x}_i - \mathbf{x}_j ||^2\) should be small). If \(A_{ij}\) is large and the distance \(|| \mathbf{x}_i - \mathbf{x}_j ||^2\) is large, the cost function will assign a high penalty, indicating that the low-dimensional embedding is distorting the relationship between these points. If points \(i\) and \(j\) are not very similar in the high-dimensional space (i.e., \(A_{ij}\) is small or zero), the cost function doesn’t care much about their distance in the low-dimensional space. Therefore, the term \(A_{ij} || \mathbf{x}_i - \mathbf{x}_j ||^2\) will be small even if \(\mathbf{x}_i\) and \(\mathbf{x}_j\) are far apart in the embedding. We can optimize this using iterative methods like gradient descend or something but there is an even more straightforward way of doing it.
Let’s define:
\(\mathbf{X} = [\mathbf{x}_1, \mathbf{x}_2, \dots, \mathbf{x}_n]^T\) as the matrix of low-dimensional coordinates.
\(D\) as the diagonal degree matrix with \(D_{ii} = \sum_j A_{ij}\).
\(A\) as the adjacency matrix.
The cost function becomes:
\[
C(\mathbf{X}) = \text{Tr}(\mathbf{X}^T D \mathbf{X}) - \text{Tr}(\mathbf{X}^T A \mathbf{X})
\]
Where \(\text{Tr}\) denotes the trace of a matrix. Using the graph Laplacian \(L = D - A\), we can express the cost function as:
\[
C(\mathbf{X}) = \text{Tr}(\mathbf{X}^T L \mathbf{X})
\]
This is the final form of the cost function in terms of the Laplacian matrix.
he Laplacian matrix \(L\) is symmetric and positive semi-definite, meaning it has real non-negative eigenvalues and can be decomposed into its eigenvectors and eigenvalues:
\[
L = U \Lambda U^T
\]
Where: - \(U\) is the matrix of eigenvectors of \(L\), - \(\Lambda\) is the diagonal matrix of eigenvalues.
The goal of spectral embedding is to minimize \(C(\mathbf{X})\). To minimize this cost, we find the eigenvectors corresponding to the smallest non-zero eigenvalues of \(L\), which provide the smoothest variation across the graph.
We can calculate this using the following code:
# Compute the degree matrixD <-diag(rowSums(graph))# Compute the Laplacian matrixL <- D - graph# Eigen decomposition of the Laplacian matrixeigen_decomp <-eigen(L)# Extract the eigenvectors corresponding to the smallest non-zero eigenvalues# The first eigenvalue corresponds to the smallest eigenvalue (which is 0), so skip it.k <-2# Number of dimensions for the embeddingembedding <- eigen_decomp$vectors[, 2:(k+1)] # Take the next k eigenvectors
We can now use this embedding instread of random initialization.
Code
# set a and ba <-1.895526b <-0.8005876# set learning ratealpha <-1# number of componentsn_components <-2# negative_sample_ratenegative_sample_rate <-5# Step 1: Initialize embeddings using spectralset.seed(10)Y <- embeddingn_epochs <-200# Step 2: Optimization loop (Epochs)# Attractive forcesedges <-which(graph !=0, arr.ind =TRUE) n_edges <-nrow(edges)for (epoch in1:n_epochs) {for (e_idx in (1:n_edges)) { i <- edges[e_idx, 1] j <- edges[e_idx, 2] p_ij_value <- graph[i, j]if(runif(1)<=p_ij_value){# Compute attractive gradient grad_attr <-update_attractive(Y[i, ], Y[j, ], a, b)# Update embeddings Y[i, ] <- Y[i, ] + alpha * grad_attr Y[j, ] <- Y[j, ] - alpha * grad_attr # Symmetric updatefor (n in1:negative_sample_rate) { k <-sample(setdiff(1:N, i), 1) # Ensure k != i grad_rep <-update_repulsive(Y[i, ], Y[k, ], a, b, 1)# Update embedding Y[i, ] <- Y[i, ] + alpha * grad_rep } } } alpha<-1*((1-epoch/n_epochs))}plot(Y)
The embedding is now much better embedding and the algorithm converges much faster. With this we wrap up the UMAP section.
3.7 Summary
UMAP, which we derived together, works by preserving both local and global structures in a more efficient and scalable way compared to t-SNE. The algorithm began by constructing a graph based on the relationships in the high-dimensional space, using the n_neighbors parameter to determine how many neighbors to consider for each data point. UMAP then converted these distances into a fuzzy topological structure, where the graph’s weights represented the probability of points being connected. This graph approximated the local manifold structure of the high-dimensional data.
For the low-dimensional space, UMAP created another graph and sought to match the two graphs by minimizing a cross-entropy loss function. This function measured the difference between the high- and low-dimensional fuzzy topological structures. The optimization process adjusted the positions of the points in the low-dimensional embedding to minimize the loss, balancing the need to preserve local details while also capturing broader global structures.
By using this approach, UMAP efficiently captured both local and global structures of the data. Its reliance on topology and manifold learning allowed for more flexible and scalable dimensionality reduction, especially for large datasets.
This overall process made UMAP an effective tool for revealing clusters, patterns, and global structures in the data, while also providing more computational efficiency than t-SNE.